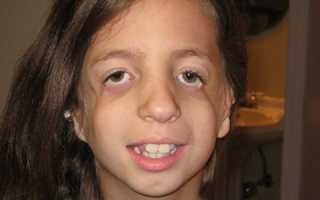
Стадии
Учитывая всю проблематичность мутаций, проявления данного недуга могут быть выражены очень ярко, или же быть незначительными.
Классифицируют такие стадии патологического процесса:
- На первом этапе видоизменения практически незаметны. Такие пациенты счастливчики, ведь они попали именно в ту категорию, которая позволяет им вести полноценный образ жизни.
- На второй стадии признаки налицо, как в прямом, так и в переносном смысле. Также могут возникнуть и сложности с функцией дыхания и функционированием других органов.
- Одной из наиболее сложных является третья стадия. Она сопровождается почти полной деформацией лица. При этом никакая терапия и пластика не будут эффективны.
Лечение синдрома Франческетти
К сожалению, необходимо констатировать тот факт, что в настоящее время медицина не готова предложить терапевтические методы лечения данной проблемы, их попросту не существует. Вся терапия направлена исключительно на паллиативную помощь. Человек, который отличается от всех остальных, сильно желает быть кому-то нужным. Поэтому в этом случае ценнее простой человеческой заботы трудно что-либо найти.

Если наблюдаются тяжёлые стадии заболевания, то следует делать операцию. Если у человека при этом есть проблемы со слухом, врачи рекомендуют использовать слуховой аппарат. Конечно, от моральной поддержки зависит очень многое. Сможет ли пациент поверить в себя? Сможет ли быть сильнее всех проблем? Довольно трудные вопросы. Это говорит о том, что психологическая помощь не может быть лишней никогда. В любой ситуации при возможности необходимо поддержать добрым словом. Вам это ничего не стоит, а больному это может открыть второе дыхание.
Синдром Тричера Коллинза: причины
Синдром Тричера Коллинза является аутосомно — доминантным заболеванием. Это значит, что дефектный ген этой болезни не связан с половой хромосомой, а значит, равносильно может появиться и у мужчин и у женщин. Кроме того, этот ген является доминантным, что означает, что при его наличии в организме, синдром Тричера Коллинза проявится в 100% случаев наличия этого гена.
Таким образом, развитие заболевания Тричера Коллинза не зависит от воздействия каких-либо вредных внешних и внутренних факторов. Можно сказать, что это заболевание уже заложено в генетический код будущего ребенка и начинает раскрываться задолго до его рождения.
Причина появления синдрома Тричера Коллинза у новорожденного ребенка кроется еще на этапе эмбриогенеза плода и процесса закладки органов. Именно в это время, а точнее на 7-й неделе развития эмбриона, происходит мутация в определенной хромосоме человеческого генетического кода – 5-й хромосоме. Эта хромосома является самой длинной в геноме человека, и именно она отвечает за синтез материала для будущего костного скелета. В результате в этой хромосоме происходит особая мутация — так называемая «нонсенс-мутация». Особенность этой мутации кроется в особенностях внутриклеточного синтеза белка. В норме такой важный процесс как биосинтез белка происходит следующим образом: цепь ДНК «перезаписывает» информацию на вспомогательную единицу – РНК. Буквально говоря РНК клонируется, записывая на себя определенную последовательность участков ДНК. Эти участки представляют собой определенную последовательность составных частей нуклеотидов, каждый из которых несет свою особую информацию. Но кроме нуклеотидов существуют и особые гены, которые получили название «стоп-кодонов». Эти гены выполняют особую функцию — в последующей сборке белка на основе РНК они в заканчивают постройку молекулы белка.
После того, как будет создана РНК с полной информацией, аналогичной информации материнской ДНК, она транспортируется в особый клеточный орган-рибосому. Именно она занимается синтезированием будущего белка — основы клеточной структуры определенных органов. РНК проходит сквозь рибосому, а точнее через ее функциональные участки. Эти участки взаимодействуют с РНК, «считывают» с нее информацию и вырабатывают белковые цепи, где каждая из них соответствует своему нуклеотиду на РНК. Но когда функциональный центр взаимодействует с описанным выше геном стоп кодоном, то он получает информацию о прекращении синтеза белка. Говоря своими словами, стоп кодоны, как бы «отрезают» от общей массы отдельные полипептидные цепи, где каждая имеет свою структуру. Позже эти цепи будут собраны в белковые молекулы.
Но при синдроме Тричера Коллинза в 5-й хромосоме, в ее гене под названием TCOF1 происходит сбой — на месте нормальных нуклеотидов, способных создать полипептидную цепь, образуется стоп-кодон. В результате, при дальнейшем синтезе происходит преждевременное окончание сборки белка, и такой белок получается дефектным. Как результат, развивается синдром гаплонедостаточности — количества образуемого белка при такой недостаточности просто недостаточно для синтезирования будущего прототипа костной структуры лицевой части черепа и последующего правильного развития из него самой костной структуры.

Как следствие этого, развивается формирование целого ряда деформаций лицевой части черепа: нарушение пропорций ее костной части, атрезия (недоразвитие) ушных раковин и наружного слухового прохода, полное или частичное нарушение формирования правильных черт лица.
Возможные осложнения и прогноз
Если заболевание не сопровождается грубыми пороками развития внутренних органов, то прогноз для жизни и здоровья ребенка в будущем благоприятен. При наличии тугоухости снижается способность к развитию речи, письма, обучению.
Необычная внешность также мешает получению навыков социального общения, так как другие дети стараются избегать сверстников с врожденными аномалиями и уродствами. В результате у ребенка может быть значительно снижена самооценка, что требует консультации со стороны психолога. Проведение ряда пластических операций позволяет частично сгладить дефекты развития.
Тугоухость часто ошибочно диагностируется как недоразвитие психики. Умственная отсталость у таких детей встречается редко, а некоторые имеют способности выше среднего
Поэтому у таких пациентов важно на ранней стадии выявлять нарушения слуха и корректировать их
Врачи рекомендуют это делать в обязательном порядке до достижения ребенком шестимесячного возраста. С 3-х месяцев он может носить аппарат для костного проведения звуковых волн, а после 3 лет возможна установка имплантата за ушами. Также очень большое значение имеют занятия с сурдопедагогом.
В первые месяцы жизни недоразвитость нижней челюсти может способствовать выпадению языка и перекрытию дыхательных путей, что является потенциально опасным для жизни ребенка. В последующем могут возникнуть затруднения в приеме пищи из-за ограниченной возможности открытия рта (в различной степени тяжести).
Синдром Тричера Коллинза
Синдром Тричера Коллинза – это страшное и довольно редкое заболевание, негативным образом влияющее на формирование костных тканей. В результате этой болезни у человека происходит черепно-лицевая деформация, которая полностью меняет его внешний вид.
Специалисты говорят о том, этот синдром в различной степени встречается примерно у одного ребенка из 50 000.
 Читайте также:
Читайте также:

Совсем недавно Йоно узнал о том, что в далекой Австралии есть маленький мальчик по имени Закери Уолтон (Zackary Walton), который страдает от такой же болезни. Месяц назад Ланкастер преодолел огромное расстояние из Англии до Австралии, чтобы встретиться с Закери.
Мать мальчика Сара Уолтон была очень рада приезду Йоно. Ей очень хотелось, чтобы её сын познакомился с человеком, у которого такая же болезнь. Ведь Йоно на своём примере показал маленькому Закери, что, несмотря на страшный недуг, можно жить полноценной жизнью.
Это знакомство стало знаменательным событием и для самого Йоно. По его словам, в детстве ему очень не хватало примера для подражания, а именно человека, у которого была та же болезнь, что и у него, но, который бы, несмотря на неё, имел бы хорошую работу и любимую девушку.
Важно, чтобы человек со страшным диагнозом не чувствовал себя одиноким. Порой это просто необходимо, чтобы человек, у которого такая же проблема, поддержал бы, сказав: “Ты можешь иметь всё, ты сможешь добиться многого, и болезнь этому не помеха”
Йоно Ланкастер и маленький Закери впервые встретились 17 ноября. На своей страничке в социальной сети мужчина опубликовал фотографии, которые подписал так: “Следующие пару дней я собираюсь провести со своим маленьким приятелем и его чудесной семьёй в прекрасном австралийском городе Аделаида”.
Стоит отметить, что Ланкастер подрабатывает в качестве модели. Однако, он не является новичком в правильном подходе к больным детям.
Маленький Закери – не единственный ребенок, которому Йоно оказывает помощь. Его страничка в Twitter подтверждает, что он является покровителем благотворительной организации “Жизнь для ребенка” (“Life For a Kid”), сотрудники которой помогают детям, чья жизнь омрачена тяжелыми болезнями.
Удивительно, но человек, которому, казалось бы, и самому нужна поддержка окружающих, обладает огромной силой воли и твердостью духа. Несмотря на редкую болезнь, которая могла бы омрачить ему жизнь, Йоно Ланкастер не чувствует себя обиженным.
Молодой мужчина живёт полноценной жизнью, в отличие от многих здоровых людей, он умеет радоваться каждому дню, принимая его как дар. Бесспорно, Йоно Ланкастер может стать примером сильного духа человека.
-
Читайте также:

Синдром Тричера Коллинза: причины болезни и лечение заболевания
Синдромом Тричера-Коллинза называют заболевание наследственного характера, характеризующееся деформацией костных структур черепа и лица. Другое название болезни – челюстно-лицевой дизостоз.
Патология возникает у одного новорожденного на 50 тысяч малышей. Подробнее о синдроме Тричера-Коллинза и жизни людей, имеющих дефекты на уровне генов, рассказано в статье.
Синдром Тричера-Коллинза: причины заболевания
Эдвард Тричер Коллинз описал основные нарушения, возникающие у больных людей, более ста лет назад. Патология получила соответствующее название. Современная медицина предпочитает использовать другие термины – мандибулофасциальный дизостоз или синдром Франческетти-Коллинза.
Основным фактором, который приводит к развитию заболевания, считается врожденная аномалия в гене 5-ой хромосомы.
Этот ген считается ответственным за кодировку белка, принимающего участие в правильном формировании костей лицевой части черепа в период внутриутробного развития ребенка. Белок называется ядрышковым фосфопротеином.
В конце первого месяца беременности лицевой отдел плода заполняется мезенхимальными тканями, которые на протяжении второго месяца уже дифференцируются в кости и соединительнотканные элементы.
Заболевание может возникнуть в случае тератогенного влияния следующих факторов:
- этанол и его производные;
- радиоактивное излучение;
- цитомегаловирус;
- токсоплазмоз;
- влияние гербицидов;
- препараты на основе ретиноевой кислоты;
- противосудорожные лекарства;
- психотропные препараты.
Симптомы патологии
Основными проявлениями синдрома Тричера-Коллинза считаются:
- опущенные углы глаз;
- недоразвитость скуловых костей с обеих сторон;
- отсутствие части тканей века;
- недоразвитость кости челюсти;
- отсутствие или малый размер раковин ушей;
- отсутствие или заращение наружных слуховых проходов врожденного характера;
- анатомические дефекты среднего отдела уха;
- «птичье лицо».
Так как височная кость является сложным элементом черепно-лицевого отдела, в некоторых случаях всю тяжесть поражений анатомо-физиологических особенностей наружного слухового прохода и внутренних структур уха оценить достаточно трудно.
-
Читайте также:

Визуальный осмотр больного человека позволяет определить отсутствие ресниц на нижнем веке, в некоторых случаях – расщелину неба. При синдроме Тричера-Коллинза возможны врожденные дефекты и нарушения работы со стороны сердца и элементов опорно-двигательного аппарата (помимо лицевого отдела).
Особенности жизни человека с синдромом Тричера-Коллинза можно увидеть в документальном фильме «Новое лицо Джулианы».
На момент одиннадцати лет ребенок перенес уже более 50 операций, но лечение еще продолжается.
Родители Джулианы имеют также старшую здоровую дочь. Они хотели еще одного ребенка, однако, побоялись его планировать из-за возможности передачи генной мутации. Взамен родители удочерили украинскую девочку, имеющую такую же генетическую болезнь, но в менее тяжелом проявлении.
Лечение синдрома Тричера-Коллинза
Это заболевание, как и множество других генетических дефектов и нарушений, лечению не поддается. Используется паллиативная терапия, которая позволяет поддержать жизнь больного на достаточном уровне.
Для коррекции слуховых функций используют слуховые аппараты, а чтобы улучшить речь, необходима помощь специалиста-логопеда.
Показан ряд и других пластических операций для лечения и восстановления анатомических дефектов.
Джон Ланкастер и синдром Тричера-Коллинза
Джон Ланкастер – парень, проживающий в Великобритании. На данный момент ему 26 лет. Биологические родители отказались от Джона сразу после его рождения, они не смогли смириться с грозным заболеванием, однако ребенка усыновила другая пара, которая смогла обеспечить его всем необходимым.
Ланкастер перенес несколько пластических операций, которые немного изменили его внешность.
На данный момент он показывает своим примером, что даже с генетическими аномалиями можно жить и радоваться жизни.
Сестра Аделины Сотниковой и синдром Тричера-Коллинза
Аделина Сотникова – российская олимпийская чемпионка по фигурному катанию. Ее младшая сестра Маша имеет генетическое заболевание, о чем стало известно недавно, после победы Аделины в Сочи.
После рождения младшей сестры родителей Аделины стали готовить в роддоме к тому, что больного синдромом Тричера-Коллинза ребенка придется оставить, но они отказались, приняв решение заботиться о дочери и бороться за ее здоровье.
Свои гонорары Аделина Сотникова тратила на лечение сестры Маши. Девушке еще предстоят хирургические вмешательства, поскольку она продолжает расти.
Последствия

Помимо внешних признаков патология себя не проявляет
Недуг очень серьезен и опасен своими осложнениями. Обычно это проблемы со слухом. Может быть недоразвит рот, что приводит к невозможному самостоятельному приему пищи. В результате зарастания носовых проходов могут происходить нарушения дыхательных функций. Также верхнее небо может быть недостаточно развитым и загромождать дыхательные ходы.
Помимо внешних признаков недуг больше никак себя не проявляет. Интеллектуальные способности больных детей абсолютно равны со здоровыми людьми. Такие пациенты полноценно развиваются, находят себя в жизни.
Многие нуждаются в квалифицированной психологической помощи. Уродства откладывают свой отпечаток на внутренних переживаниях личности, порождают множество комплексов, делают людей замкнутыми.
Такой диагноз носят пожизненно. Есть только один выход — сделать все возможные операции и жить полноценно, не обращая внимания на свои особенности.
Диагностика у новорождённого
Определение заболевания у новорожденного обычно не вызывает затруднений и производится по внешним признакам. Генетический анализ для дополнительного подтверждения диагноза производится по венозной крови, таким же способом, как во время беременности.
Стоимость такого анализа порядка 60 тыс. руб. (вместе с заключением врача-генетика). При выявлении отклонения также рекомендуется сделать исследование для обоих родителей, братьев и сестер.
Дополнительно проводятся следующие обследования:
- определение остроты слуха, методика проведения зависит от возраста ребенка: регистрация вызванных слуховых потенциалов (подача акустических сигналов и регистрация электроэнцефалограммы), речевая аудиометрия (произнесение слов и фраз с различной громкостью), тональная пороговая аудиометрия (проводится с помощью аудиометров, результатом является графическое изображение слухового порога);
- рентгенография черепа;
- компьютерная томография височных костей по достижении возраста 3 лет для подготовки к хирургической операции;
- консультация у профильных специалистов (отоларинголога, окулиста, кардиолога, хирурга и других).
Данное заболевание требует дифференциальной диагностики с такими генетическими патологиями, как:
- Синдром Нагера. Для этой болезни также характерен антимонголоидный разрез глаз, недоразвитие нижней челюсти и патологические изменения в органах слуха и зрения. Кроме этого, у больных детей имеются грубые нарушения в развитии конечностей – укорочение и искривление предплечья, фаланг и локтевых костей, отсутствие пальцев и сращение костей рук.
- Синдром Гольденхара. Неправильное развитие затрагивает не только лицо и череп, но и нервную, сердечно-сосудистую, мочевыделительную, пищеварительную системы, у многих больных отмечается умственная отсталость. Как правило, данная аномалия имеет односторонний характер (70% больных).
Диагностика
Диагноз синдрома Тришера Коллиса основан на клинических и рентгенологических данных и, кроме того, используется несколько дополнительных генетических тестов (Genetics Home Reference, 2016).
В случае клинического диагноза проводится подробное физическое и неврологическое обследование, чтобы уточнить их. Обычно этот процесс выполняется на основе диагностических критериев заболевания..
Одним из наиболее часто используемых тестов на этом этапе оценки являются рентгеновские снимки, они могут предоставить нам информацию о наличии / отсутствии черепно-лицевых пороков развития (Национальная организация по редким заболеваниям, 2013 г.).
Хотя некоторые черты лица можно наблюдать непосредственно, рентген дает точную и точную информацию о развитии челюстных костей, развитии черепа или развитии дополнительных пороков развития (Национальная организация редких заболеваний, 2013).
Кроме того, в тех случаях, когда физические признаки все еще очень незначительны или в которых необходимо подтвердить диагноз, можно использовать различные генетические тесты для подтверждения наличия мутаций в генах TCOF1, POLR1C и POLR1D (Национальная организация по редким заболеваниям). , 2013).
Кроме того, когда есть семейный анамнез синдрома Трешира Коллинза, можно выполнить пренатальную диагностику. Посредством амниоцентеза мы можем исследовать генетический материал эмбриона.
Диагностика во время беременности
Диагностика заболевания плода в период его вынашивания производится 2 способами: с помощью ультразвукового исследования, на котором выявляются грубые челюстно-лицевые нарушения, и молекулярного исследования образцов биологического материала.
Молекулярно-генетическая диагностика позволяет выявить изменения в ответственном гене путем прямого определения аминокислотной последовательности, которое производится автоматически.
В качестве биологического материала используются:
- образцы ворсин внешней оболочки зародыша, если срок беременности находится в пределах 8-14 недель;
- околоплодные воды, если женщина беременна на сроке 16-21 недель.
Стоимость исследования составляет порядка 30 тыс. руб., срок выполнения около 1 месяца, цена поиска мутации у родственника – 2800-3500 тыс. руб.
Если в семье уже имелись подобные случаи генетических нарушений, то настоятельно рекомендуется сделать диагностику перед подготовкой к беременности (стоимость исследования – 8-10 тыс. руб.). Такое исследование является единственным способом профилактики возникновения данной патологии у ребенка.
Заключение
Синдром Коллинза встречается довольно редко, однако является серьёзным заболеванием. Причиной данной болезни является наследственность, спонтанная мутация генов. Врачи во время обследований беременной женщины начинают проверку на наличие деформации лицевой части плода. Если исследование показало, что болезнь существует, специалист обязан составить курс терапии, который максимально облегчит жизнь человеку.
Синдром Тричера Коллинза – генетическое аутосомно-доминантное заболевание, характеризующееся неправильным развитием костей лицевого скелета. Черепно-лицевые деформации происходят на ранних сроках беременности в результате генетической мутации. Диагностировать внутриутробные нарушения можно во или сразу после рождения ребёнка. Частота фиксируемых случаев рождения младенцев с синдромом Тричера Коллинза в различных источниках указывается разная – от 1 случая на 10-50 тысяч родов. Заболевание считается малоизученным и пожизненным.