Основные характеристики
Особенности ключично-черепной дисплазии сильно различаются от одного человека к другому, однако наиболее распространенные из них включают:
- Задержка закрытия мягких мест у малыша;
- выступающий подбородок и лоб;
- Очень широкий нос;
- Крыша рта выше нормы;
- Укороченные или отсутствующие ключицы;
- Узкие и очень гибкие плечи;
- Замедленный рост зубов.
Кроме того, дисплазия также может поражать позвоночник, и в этих случаях могут возникнуть другие проблемы, такие как, например, сколиоз и низкий рост. Точно так же изменение костей лица может также привести к модификации пазух, что может привести к более частым приступам синусита у ребенка с ключично-черепной дисплазией.
Челюстно-черепной дизостоз
Челюстно-черепной Дизостоз (синдром Петерс — Хевельса) — гипоплазия верхней челюсти, скуловых дуг, открытый прикус, прогения (выстояние нижней челюсти), укорочение переднего отдела основания черепа. Аномалия наследуется по доминантному типу.
Существуют другие формы черепных Д.: синдромы Гегенхара, Робена, Франсуа и др. Внешний вид больных с различными формами Д. характерен. Д. сохраняется всю жизнь, не поддается оперативной коррекции, почти не требует дифференциальной диагностики с другими заболеваниями. В сомнительных случаях важным диагностическим методом является рентгенологическое исследование.
Различают так наз. неполные типы перечисленных Д., когда имеют место не все характеризующие их симптомы. Отдельные признаки могут комбинироваться в различных сочетаниях, составляя как бы промежуточные типы Д.
Прогноз для жизни благоприятный.
Люди с синдромом Тричера Коллинза
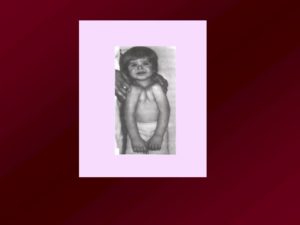
Синдром Тричера – генетическое заболевание, на возникновение которого в большинстве случаев не влияют никакие внешние или внутренние факторы.
Можно сказать, что патология изначально заложена в аминокислотный код будущего ребенка и начинает проявляться задолго до его рождения. Научно доказано, что спонтанные изменения в структуре ДНК (генные мутации) у лиц, имеющих синдром, возникают в 5 хромосоме.
Последняя является самой длинной нуклеотидной структурой в геноме человека и отвечает за производство материала для скелета плода.
 Читайте также:
Читайте также:

Происходят мутации по причине сбоя внутриклеточного синтеза белка. В результате чего развивается синдром гаплонедостаточности. Последний характеризуется нехваткой белка, необходимого для правильного развития лицевой части черепа. При всем этом следует знать, что болезнь Тричера-Коллинза имеет аутосомно-доминантный, реже – аутосомно-рецессивный характер.
- этанол и его производные;
- цитомегаловирус;
- радиоактивное излучение;
- токсоплазмоз;
- прием противосудорожных и психотропных препаратов, лекарств с ретиноевой кислотой.
Диагностика
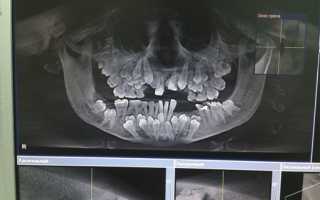
Диагностика ключично-черепного дизостоза основана на клинических симптомах и рентгенологических исследованиях, которые включают изображения черепа, грудной клетки, таза и рук.
Главный рентгенологический симптомом — дефекты ключиц. Обычно отсутствует наружный (акромиальный) конец ключицы, в то время как внутренний (грудинный) конец присутвует. Но иногда ключица состоит из двух фрагментов.
Полное отсутствие ключицы встречается редко.
При молекулярно-генетическом исследовании обнаруживаются мутации гена RUNX2 у 60 % −70 % людей с диагнозом ключично-черепной дизостоз.
Постнатальная диагностика проводится на основании имеющихся клинических проявлений. При полной экспрессивности синдрома Тричера вопросов, как правило, не возникает, чего нельзя сказать, когда обнаруживаются незначительные признаки данной патологии. В этом случае проводится комплексная диагностика состояния, включающая следующие исследования:
- оценку и мониторинг эффективности кормления;
- аудиологическое тестирование слуха;
- рентгеноскопию черепно-лицевой дисморфологии;
- пантомографию;
- КТ или МРТ головного мозга.
Аналогичные методы исследования применяются, когда необходимо провести дифференциальную диагностику для того, чтобы распознать неярко выраженные проявления болезни Тричера-Коллинза и отличить их от признаков других патологических состояний. Так, в большинстве случаев специалисты назначают дополнительные инструментальные исследования для дифференциации указанного недуга с синдромами Гольденхара (гемифациальной микросомии), Нагера.
Какие мутации приводят к развитию синдрома Тричера Коллинза
Чаще всего при синдроме Тричера Коллинза происходит мутация в генах TCOF1, POLR1C и POLR1D. При этом изменения в гене TCOF1 обнаруживается в 93% всех случаев постановки этого диагноза.
Мутация в генах POLR1C и POLR1D выявляется довольно редко. Именно это является причиной развития синдрома Тричера Коллинза.
-
Читайте также:

Если же нарушений в этих генах нет, но заболевание присутствует, то причину его можно считать неизвестной.
Изменения в вышеперечисленных генах сокращают общее количество производимых молекул. Предполагают, что это приводит к самоуничтожению некоторых клеток, которые отвечают за развитие тканей лица и черепа. Всё это ещё во время формирования плода приводит к тому, что в формировании лица есть некоторые проблемы, которые могут быть как едва заметными, так и сильно выраженными.
Болезнь Тричера-Коллинза имеет три стадии. На начальном этапе его развития наблюдается незначительная гипоплазия лицевых костей. Вторая стадия характеризуется деформацией и недоразвитостью слуховых проходов, маленькой нижней челюстью, аномалиями глазной щели, что прослеживается практически на всех фото пациентов с синдромом.
История
Первый случай отклонений в развитии ключиц был описан М. Мартин в 1765 году. Случай, связанный с отклонениями в развитии ключиц и костей черепа был рассмотрен в 1861 году Шейтхауэром.
В 1897 году Мари и Сентон более подробно рассмотрели это заболевание, указали на семейный характер патологии, и дали название dyostosis cleido-cranialis, поскольку соотносили его с дефектами костей черепа и ключиц.
Гессе в 1926 году описал отклонения в развитии зубов и челюстей и связал его с ключично-черепным дизостозом.
Ключично-черепная дисплазия
Для этого вида дисплазии типична разная степень гипоплазии мембранозных костей. Она наследуется по аутосомно-доминантному типу. У больных гипоплазируются грудинные концы ключиц и отростки крестцовых позвонков, поздно закрывается передний родничок в виде образования многочисленных червеобразных косточек. Вследствие гипоплазии ключиц плечи опущены и выдвинуты вперед. Характерны выступающие лобные бугры, разболтанность суставов, приводящая к вальгусному искривлению коленного сустава, и аномалии развития зубов. Появление молочных зубов задерживается, их число зачастую уменьшено. Постоянные зубы также появляются позднее, расположены неправильно, деформированы или гипоплазированы. Возможно пропорциональное укорочение роста.Рентгенологически отмечается разной степени гипоплазия ключиц и лопаточных костей. Окостенение лобковых и седалищных костей замедлено, лобковый симфиз расширен.
Черепно-лицевой дизостоз
Черепно-лицевой Дизостоз (синдром Крузона, гипертелоризм) — недоразвитие костей черепа, мозга и верхней челюсти в сочетании с преждевременным закрытием черепных швов, экзофтальмом, косоглазием, нистагмом, расстройством зрения. Лоб в области переносицы бугрист, глаза широко расставлены, нос своеобразной крючковидной формы («клюв попугая»), гипоплазия верхней челюсти, псевдопрогения; в резко выраженных случаях наблюдается снижение умственного развития. Наследуется по доминантному типу.
Рентгенологически выявляются изменения черепа. На первый план выступает характерная деконфигурация головы и нарушение нормальных соотношений между мозговым и лицевым черепом: первый уменьшен в размерах, имеет почти шаровидную форму, швы заращены, усилены пальцевые вдавления. Кости свода черепа истончены, несколько выпячиваются кнаружи в области переднего родничка. Основание черепа укорочено и углублено, область турецкого седла сужена, глазницы уплощены.
-
Читайте также:

Кости лицевого черепа малы: верхняя челюсть и носовые кости недоразвиты, нижняя челюсть значительно выдается вперед, в силу чего образуется резкий прогиб носа внутрь.
Рис. 3. Ребенок с челюстно-лицевым дизостозом. Характерны широкие косо расположенные глазные щели, нарушение развития зубов.
Черепно-ключичный дизостоз: причины появления и лечение заболевания
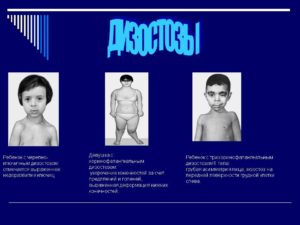
Основными симптомами наличия данной патологии являются:
- отсутствие или недоразвитие обеих или одной из ключиц, из-за чего плечевой пояс сильно сужен, а надплечья опущены. По этой же причине в плечевых суставах заметна чрезмерная подвижность;
- задержка окостенения родничков, при этом в некоторых случаях формируются дополнительные костные включения. В некоторых случаях большой родничок так и не закрывается на протяжении всей жизни человека.;
- присутствие нарушений в формировании корней зубов, а также задержки при прорезывании постоянных и молочных зубов. Иногда у людей с этой патологией молочные зубы не меняются до 30 лет, но при этом отмечается наличие сверхкомплектных зубов.
В большинстве случаев отмечаются также такие симптомы, как брахицефалия, низкий рост (в сравнении с родственниками), гипертелоризм, недоразвитие костей таза, выдающийся вперед и высокий лоб.
У пациентов с классической формой этой патологии обычно нормальный уровень интеллекта, но существует ряд других медицинских проблем. Так, такие люди подвержены рецидивам и осложнениям инфекций ушей, дыхательных путей, у них рано появляются проблемы с суставами и развивается остеопороз, а у детей отмечают легкую степень моторной задержки.
Челюстно-лицевой дизостоз – симптомы
Выявить челюстно-лицевой дизостоз можно уже при первом осмотре пациента:
- У таких детей отмечается выраженная недоразвитость скуловых костей, челюсти.
- Ушные раковины полностью не сформированы: уши у ребенка имеют малый размер, а слуховой канал недоразвит.
- Разрез глаз заметно сужается.
- Тип и выраженность нарушения может варьироваться, однако всегда происходит сбой в работе слухового аппарата, дыхательной системы.
Самым ярким симптом, сопровождающим болезнь Тричера-Коллинза, является изменение нормальной формы глаз. Глазная щель сужается, в результате чего глазные яблоки опускаются. Возможны и другие проявления синдрома Тричера-Коллинза:
- дефект мягкой ткани ротовой полости;
- впалый подбородок;
- нарушение слуха;
- нарушение прикуса;
- расщепление верхнего неба;
- изменение прикуса.
Синдром Тричера-Коллинза – степени
Как отмечалось выше, патология развивается постепенно, прогрессируя со временем. Синдром Тричера-Коллинза в начальной степени характеризуется незначительной гипоплазией лицевых костей. Диагностируется уменьшение в размерах скуловых костей, из-за чего лицо выглядит слегка вытянутым. Такое изменение именуют первой степенью заболевания.
-
Читайте также:

Челюстно-лицевой дизостоз или синдром Тричера-Коллинза второй степени сопровождается недоразвитием слуховых проходов. Нижняя челюсть значительно меньшего размера, глазная щель сужается, что приводит к нарушению зрения. При тяжелой форме лицо практически отсутствует. Нос, скуловые кости, верхняя и нижняя челюсти деформируются настолько, что пациент становится неузнаваемым для окружающих.
Синдром Тричера-Коллинза – прогноз
Синдром Тричера-Коллинза является тяжелым испытанием для пациентов. Прогноз полностью зависит от степени выраженности деформации лицевых костей, количества сопутствующих клинических нарушений. В большинстве случаев патологии прогноз благоприятный. Но дети с данным заболеванием нередко сталкиваются с трудностями социальной адаптации, что негативно отражается на общем самочувствии.
Диагностика
Диагностика ключично-черепного дизостоза основана на клинических симптомах и рентгенологических исследованиях, которые включают изображения черепа, грудной клетки, таза и рук.
Главный рентгенологический симптомом – дефекты ключиц. Обычно отсутствует наружный (акромиальный) конец ключицы, в то время как внутренний (грудинный) конец присутвует. Но иногда ключица состоит из двух фрагментов. Полное отсутвие ключицы встречается редко.
При молекулярно-генетическом исследовании обнаруживаются мутации гена RUNX2 у 60% -70% людей с диагнозом ключично-черепной дизостоз.